在生物物理学、电化学以及催化反应器设计中,研究人员和工程师会利用包含气-固和液-固界面的固体表面的特殊化学与物理性质。本文将讨论简单表面上的表面反应动力学的基础知识,以及如何用 COMSOL Multiphysics® 软件模拟表面反应。在后续博客中,我们将探讨如何描述均质多孔介质表面的质量传递和反应动力学。
表面为何特殊?
表面是指 多相反应 发生的位置。多相反应涉及多个相,例如催化转化器中固体表面的氮氧化物的催化还原反应。多相反应过程只能发生在不同相界面上,因为相是指系统中不同的不混溶成分。
表面也是发生吸附的位置,吸附是相邻气相或液相中的分子通过范德华力(物理吸附)等分子间作用力,或直接化学键(化学吸附)在表面集中的过程。与游离气体或溶液中的分子相比,吸附可以使气相和液相分子花更多的时间彼此靠近(即分子附着在固体表面)。此外,化学吸附可以降低破坏吸附分子中化学键所需的活化能,从而使吸附的化学物质之间的反应能够通过与自由相中不同的机理进行。这两点是固体表面能够催化反应的重要原因。
表面反应动力学
均相反应的速率可以根据单位体积和单位时间内反应的物质的摩尔数来测量,其反应速率的单位为 mol m-3 s-1。相比之下,多相反应的速率取决于可发生反应的单位面积,因此反应速率的单位表示为 mol m-2s-1 ,两者都是摩尔通量的单位。在 COMSOL Multiphysics 中建立化学模型时,添加 通量 边界条件是指定多相反应的直接方法。
我们可以想象为反应物的通量“进入”表面,相应地,生成物的通量“离开”表面。下图显示了测得的进入催化表面反应物的通量。
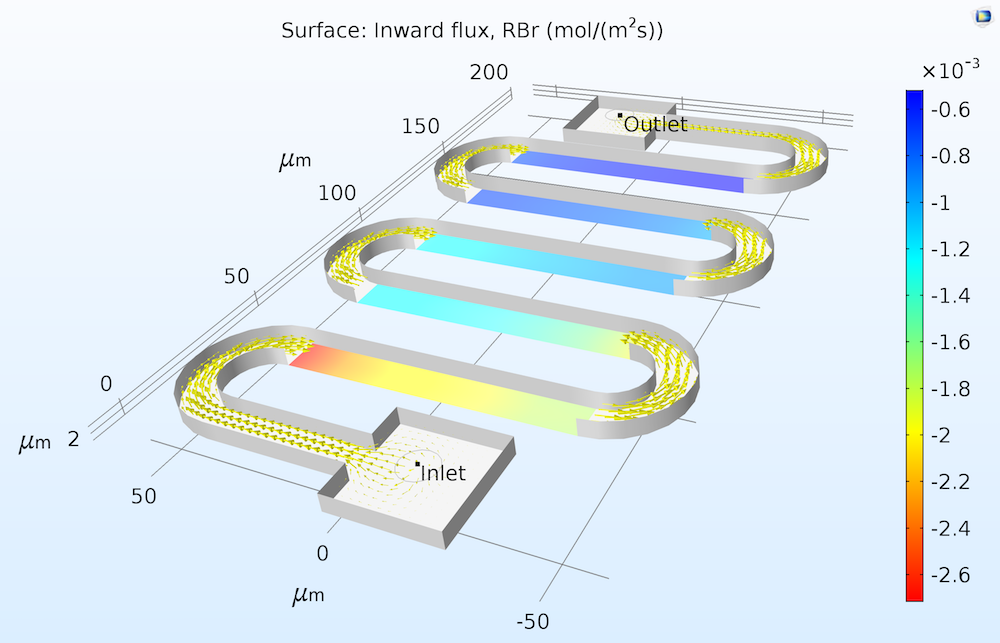
微反应器中系列催化表面上液相反应物 RBr 的通量。由于反应消耗 RBr 反应物,因此溶液的向内通量为负。
根据速率定律,表面反应速率是由反应物和生成物浓度以及其他局部性质(如温度或压力)表示的函数。大多数均相反应中的速率定律也适用于多相反应。如果您想了解一些不同的动力学速率定律,请阅读这篇关于化学动力学的介绍的博客。
以多相反应速率定律为例,一级反应消耗物质 A 可以设置 A 的向内通量,如下所示:
其中,NA 是通量 (mol m-2 s-1),cA 是表面上反应物的浓度, 不过该浓度是在相邻相中测量的(mol m-3)。
因此,一级速率常数的单位为 m s-1,表示化学物质因反应而进入边界的有效“速度”。包括电化学过程在内的表面过程的速率常数很容易求出,其单位用速度表示。
蛇形微反应器中的烃脱卤案例模型是一个模拟表面过程的示例。在这个模型中,两个相互竞争的表面反应均在特定的催化表面上发生,该表面用稀物质传递 接口中的 通量 边界条件表示。这两个反应过程的速率分别由具有不同活化能的阿累尼乌斯动力学控制。与氢化反应相比,相互竞争的二聚反应具有更高的活化能,因此随着温度的升高,反应加速越快。该模型可以预测产物比率对温度的依赖性,并将结果与实验数据进行比较。
下图显示了包含 通量 边界条件的模型开发器的一部分。如您所见,该条件特别适用于发生反应的催化表面。反应速率通过耦合的 化学 接口中指定的动力学机制定义。有关如何使用此接口模拟表面反应的更多详细信息,请参阅下一节内容。

显示了 通量 边界条件的设置的模型开发器,,用于定义由于多相反应导致的催化表面上三种溶质的通量。
表面吸附和传递
在吸附过程中,化学反应物进入表面的通量与化学反应物离开表面的通量不平衡。吸附的反应物的表面浓度(mol m-2)不断发生变化。以从气相中吸附化学物质的情况为例,我们可以将这一过程用方程的形式写出:A(g) A(ads)。
质量守恒可以表示为:
这是表面上的输运方程。Nads 表示由于表面扩散等过程而与表面相切的吸附物质的通量。在大多数情况下,这种表面扩散近似为零。在方程右边,通量作为一个反应项为吸附的物质施加质量源或质量汇。
COMSOL Multiphysics 中 表面反应 接口可实现此方程。该接口通常可以与 稀物质传递 或 浓物质传递 接口耦合,用于相邻气相或液相中的质量传递。要查看这个接口的使用示例,请阅读这篇关于蛋白质吸附模拟的博客文章。
这个示例模拟了一段离子交换柱,用于预测从水溶液流到活性表面上的两种蛋白质的摄取速率。模型的几何结构中明确包含球形离子交换珠,其表面的边界作为发生吸附反应的位置。该模型包括4个接口:
- 化学 接口,用于模拟化学机理
- 稀物质传递 接口,用于模拟流动中的溶解物质浓度(包括多相反应)
- 表面反应 接口,用于模拟离子交换珠表面吸附物质的表面浓度
- 层流 接口,用于预测水流中的速度场,从而获得对流对溶解物质传递的贡献
下面为典型的仿真绘图,描述了蛋白质暴露在水流中 30s 后的吸附程度。
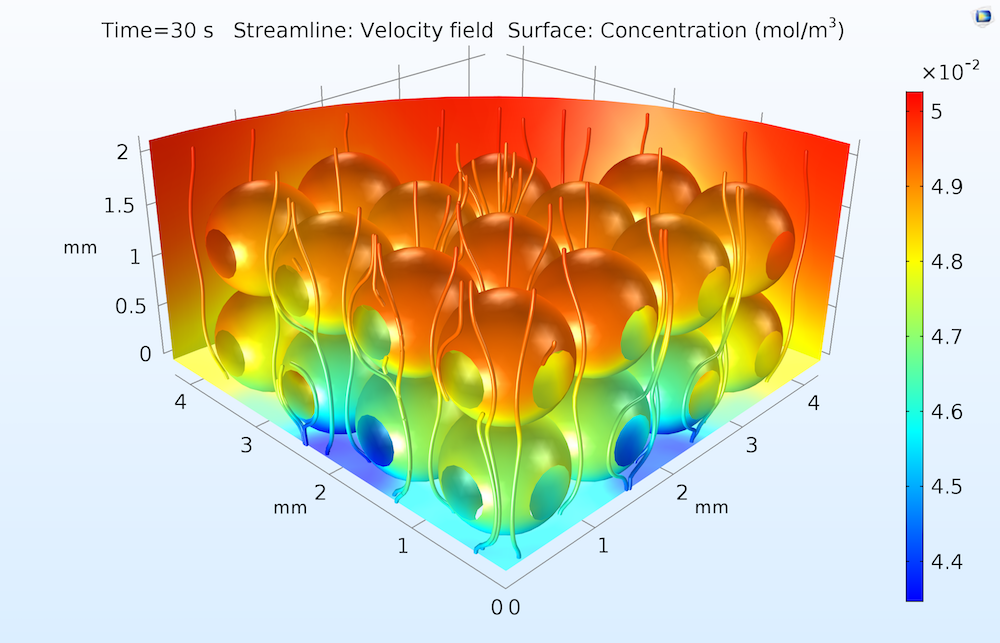
离子交换柱中吸附蛋白质暴露于含有溶解蛋白质的水流中 30 s 后的表面浓度。离子交换柱的上部(流入)面的浓度更高,其中催化表面更容易接近水流中溶解的蛋白质。
请注意,在这个示例中,吸附物质的表面扩散率设为零。因此,吸附物质浓度在任何点的变化率仅取决于局部吸附和解吸通量。动态吸附与解吸过程之间的动态平衡通常用一种被称为 吸附等温线(如 Langmuir 等温线)的数学表达式来描述。在后续即将发布的本系列博客中,我们将更详细地讨论等温线的选择和实现。
在反应工程 和化学 接口中定义表面反应
在使用 反应工程 接口描述完全混合反应器的特性时,您可以通过向反应机制中添加任何表面结合的物质来包含表面反应。表面结合的物质由物质名称末尾的后缀(ads) 表示。我们可以认为涉及表面物质的任何反应都发生在表面,因此这种反应的反应速率以 mol m-2 s-1 测量。
反应工程 接口中的 生成空间依赖性模型 选项创建了一个 化学 接口,用于存储与 稀物质传递 接口或用于模拟本体相的另一个化学物质传递接口相同。默认情况下不会跟踪吸附物质的浓度,但您可以添加额外的 表面反应 接口进行跟踪。
有两个选项可用于描述表面反应。假设这两个选项被应用于将在空间依赖性模型的几何结构中明确解析的边界,则 通量条件将被添加到化学物质传递接口来描述多相反应过程。
表面上的快速反应也可以借助 表面平衡反应 特征来描述。根据反应的平衡常数,这限制了界面上反应物和生成物的浓度以保持一定的比率。下面的屏幕截图显示了蛋白质吸附示例中的模型开发器设置。
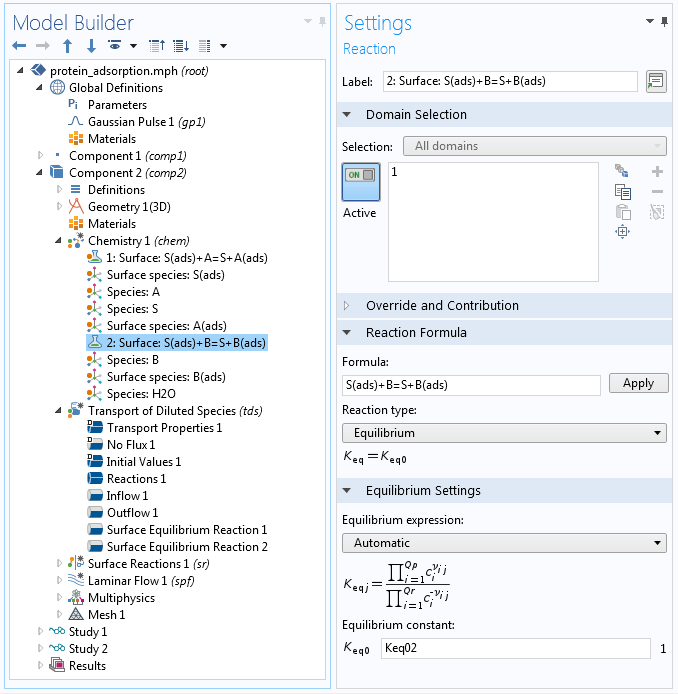
通过 化学 接口显示动力学机制规范内的表面反应设置的模型开发器。假设反应非常快速,以便根据指定的平衡常数保持平衡。
表面反应建模总结
这篇博客,我们介绍了一些表面反应的基本动力学理论,以及表面上物质的浓度与相邻本体相(气相或液相)中相同化学物质的浓度之间的质量守恒定律。使用COMSOL Multiphysics 中的 通量 边界条件和 表面反应 接口可以将这些反应添加到化学模型中。到目前为止,我们只考虑了简单形状的边界,以便将它们直接包含在几何结构中,从而定义表面反应的位置。
敬请关注本系列博客的后续文章,我们将讨论如何利用均化作用来近似具有复杂表面几何结构的多孔介质表面反应的理论。
更多资源
- 阅读 COMSOL 博客,了解更多关于化学反应建模的知识
评论 (25)
涛 商
2023-11-27速率常数的单位是1/s,而我的反应更符合二级反应,应该怎办?
越 赵
2023-11-28 COMSOL 员工您好,您可以在化学接口的表面反应中设置表面总反应级数,然后在反应速率中设置合理的反应速率即可。
永超 隽
2024-03-02comsol的工程师你好,以上蛇形反应流道和蛋白质吸附这两个案例都是稀物质传递,我想问一下,我在做浓物质传递时没有稀物质传递的“表面反应”这个节点,但是浓物质传递中多了两个默认节点“通量”和“通量不连续”,我有点不太理解这两个节点的意思,可以方便解释一下吗?除此之外,浓物质传递物理场有个“表面平衡反应”节点,这个和稀物质中的“表面反应”是一个意思吗,我看设置窗口中的方程也不一样、设置也不一样,有何异同。望收到回复,谢谢
越 赵
2024-05-07 COMSOL 员工您好!
1.表面反应是一个接口,而不是在浓物质传递的子节点中选择;2.通量用于设置外边界处物质的通量,通量不连续用于内部边界,来设置内部边界上通量的阶跃;3.表面平衡反应比稀物质传递中的表面反应要复杂一些,稀物质传递中的表面反应只需要设置反应速率即可,而浓物质传递中的表面平衡反应是基于平衡常数来计算反应的通量。具体建议您参考COMSOL帮助手册:C:\Program Files\COMSOL\COMSOL62\Multiphysics\doc\pdf\Chemical_Reaction_Engineering_Module\ChemicalReactionEngineeringModuleUsersGuide.pdf,帮助手册中有对每个节点的详细介绍。
旭 杨
2024-05-06您好,我想请教一下蛋白质吸附中的A+S(ads)=A(ads)+S后续需要给A和S一个摩尔质量和密度,这两个参数是用来定义A和S的吧?那么密度应该用物质本身的密度还是溶于水的密度,是否与浓度有关?
越 赵
2024-05-07 COMSOL 员工您好,摩尔质量和密度是用来定义A物质和S物质的属性的。密度是物质本身的密度,您可以在该案例的参数中看到rho_p是蛋白质A密度,rho_S是S的密度。
旭 杨
2024-05-08好勒,感谢感谢
奇 董
2024-05-11comsol的工程师你好,我想模拟酸性溶液(硫酸)在单一岩石(石灰岩,CaSO4)裂缝中的流动和对岩石裂缝面的腐蚀行为,需要用到哪些模块呢?有类似的案例吗?
Xiaohan Jiang
2024-05-13 COMSOL 员工推荐使用 “多孔介质中的稀物质传递”,该物理场中包含 “内表面-裂隙” 这个特征,不过暂时案例库中并没有对应的演示案例。
海胜 崔
2024-08-08工程师您好,在均质多孔介质的表面进行化学反应,需要使用哪些物理场?或者有相关案例吗?
Xiaohan Jiang
2024-08-09 COMSOL 员工一般是通过两个方式来实现的:首先是类似当前这个蛋白质吸附案例,直接将多孔介质流道微观结构在模型中绘制出来,直接利用表面反应接口计算;若所模拟的是个宏观结构,无法将大量的微观流道绘制出来,可使用多孔介质稀物质传递中的 “多空催化剂” 特征的子节点 “吸附” 功能来模拟,其中就有Langmuir、BET 等常用的吸附模型,请参考:https://cn.comsol.com/model/porous-catalytic-reactor-with-injection-needle-25。
Tianhua Ju
2024-08-28你好,模拟电解槽电解过程中金属在电极上的沉积过程也是需要添加化学反应接口的吗?谢谢!
Yi Fan Wang
2024-08-29 COMSOL 员工建议您可以直接使用通过电化学接口,您可以参阅视频“https://cn.comsol.com/video-training/electrodeposition-simulation-part-1”。
Tianhua Ju
2024-08-29请问添加了表面反应接口,执行计算时总提醒sr.d变量未定义,这个变量d到底是什么?谢谢🌹🌹🌹
错误提示:
未定义变量。
– 变量: comp1.sr.d
– 几何: geom1
Yi Fan Wang
2024-08-30 COMSOL 员工建议您通过技术支持渠道,把模型发过来,以便帮您更好的解决问题。
Tianhua Ju
2024-08-31谢谢!重新画网格解决了,虽然不知道原因。
培轩 严
2024-09-14comsol的工程师,您好,我想模拟水在容器内与活性金属的化学反应过程,可以用到哪些模块,有类似的案例吗?期待您的回复。谢谢
越 赵
2024-09-20 COMSOL 员工您好,您应该使用COMSOL的化学反应工程模块来处理该问题,如果还需要研究传热或者产生氢气后的两相流问题,您可能还需要传热模块和CFD模块来处理此类问题,目前没有类似的案例。
洪锐 贺
2025-02-23如果我想做一个电极孔道内物质的扩散模型,需要耦合电化学和稀物质传递的物理场,目的是观察在电极孔道内物质A从右侧通入,到达电极表面得电子还原,生成物质B,物质B又向右侧扩散出去的模型,对于这样的一个可以场,应该怎么设置其电极表面哪,对于这个反应又该怎么定义,需要在添加“化学”接口吗?
Jiacong Cao
2025-05-14请问如何在自由液面处施加一个随液面与基底间距离变化的压力?
Jun Leng
2025-05-27 COMSOL 员工您好!可以在流体流动如层流接口下添加自由表面边界条件,并在自由表面外压设置中输入与距离相关的表达式即可。
鲲鹏 高
2025-07-12工程师您好,我想对催化剂种类、光照条件对光热催化甲烷二氧化碳干重整效率的影响进行研究,请问需要用到哪些模块,有类似的案例嘛?期待您的答复。
育新 陈
2025-07-15 COMSOL 员工您好!
对于您描述的场景可以使用化学反应工程模块来建模,如果反应过程还涉及复杂的传热或多相流问题,可能还需要传热模块和CFD模块。这里有两个气相催化反应的案例供参考:
https://cn.comsol.com/model/porous-catalytic-reactor-with-injection-needle-25
https://cn.comsol.com/model/carbon-deposition-in-heterogeneous-catalysis-1968
三 张
2025-10-26comsol的工程师您好,我想研究二氧化碳-水-页岩(多孔介质)反应过程中的离子浓度的变化,请问涉及到哪些模块。
Haoze Wang
2025-10-27 COMSOL 员工您好!如果将多孔结构完整画出来进行仿真,涉及CFD和化学反应工程模块;如果采用均质化方法模拟多孔介质,涉及地下水流和化学反应工程模块。